Linfoma de Hodgkin
I.
INTRODUCCIÓN
El linfoma de Hodgkin (HL), uno
de los tipos de cáncer más curables, recibió su nombre de Thomas Hodgkin, un
patólogo británico. En 1832, el Dr. Hodgkin describió varios casos de personas
con síntomas de un cáncer que afecta los ganglios linfáticos. La enfermedad se
llamó “enfermedad de Hodgkin” durante aproximadamente 170 años. Su nombre se
cambió oficialmente a “linfoma de Hodgkin” a fines del siglo XX, cuando se hizo
evidente que la enfermedad era consecuencia de una lesión en el ADN de un
linfocito. El daño al ADN se adquiere; no se hereda. El ADN alterado en el
linfocito produce un cambio canceroso que, si no se trata, tiene como resultado
una proliferación descontrolada de los linfocitos cancerosos. La acumulación de
los linfocitos cancerosos produce las masas tumorales que se encuentran en los
ganglios linfáticos y otros lugares del cuerpo. Las características célulares fueron
reconocidas por Sternberg en 1898 y por Reed en 1902.
La enfermedad de Hodgkin tiene
con frecuencia un comienzo ganglionar y se extiende inicialmente a través del
sistema linfático y con posterioridad por vía sanguínea. Este tipo de extensión
y la disponibilidad de tratamientos eficaces determinan que sea curable el
75-80% de los pacientes. En la actualidad se desconoce la causa y el mecanismo
directo de esta enfermedad, aunque existen datos sobre la presencia del virusde Epstein-Barr sobre fenómenos de inmunodepresión con asociación de virus y
VIH, y ciertas anomalías citogenéticas, así como la asociación de enfermedad de
Hodgkin con otros virus.
La enfermedad de Hodgkin es poco
frecuente. Representa el 1% de todas las neoplasias malignas e incide con una
frecuencia de 3-3,5 nuevos casos por 100.000 habitantes y año. La curva de
incidencia según la edad es típicamente bimodal, con un pico en los adultos
jóvenes entre 15 y 35 años y otro después de los 55 años. El pico se desplaza a
edades tanto más juveniles cuanto menor es el nivel socioeconómico del área
estudiada.
En conjunto hay un predominio
evidente en los varones, con cifras proporcionales de hasta 1,5/1
aproximadamente. Con todo, en los adultos jóvenes la incidencia se iguala
prácticamente en ambos sexos, al ser en esta edad más frecuente la variedad
esclerosis nodular que predomina en el sexo femenino.

II. ESTADIOS DE LA ENFERMEDAD
Para determinar el estadio para
el linfoma
Hodking y no Hodking se utiliza la clasificación Ann Arbor, excepto
el linfoma cutáneo. Hay 4 estadios para clasificar esta enfermedad. Cada
estadio se subdivide: A: No sintomática y B: Si tiene fiebre, pérdida de peso,
sudoración nocturna.
Cuando presentan extensión
extralinfática se clasifican con la letra E, si afectan al bazo con la letra S,
Hígado H, Pleura P, Pulmón L, Hueso O, Médula ósea M, Piel D, Si el estadio es
clínico se antepone las letras CS, Obtenido por procedimiento quirúrgico como y
la laparotomía se antepone PS.
Estadio I:
Limitado a una
región ganglionar linfática anatómica. Afección de una sola región ganglionar
(I), o afección localizada de un solo órgano, o localización extralinfática (IE).
- Ganglio, Anillo
de Waldeyer, Timo o Bazo
- Raro en
Linfoma Hodking
- Estadios
clínicos cS.
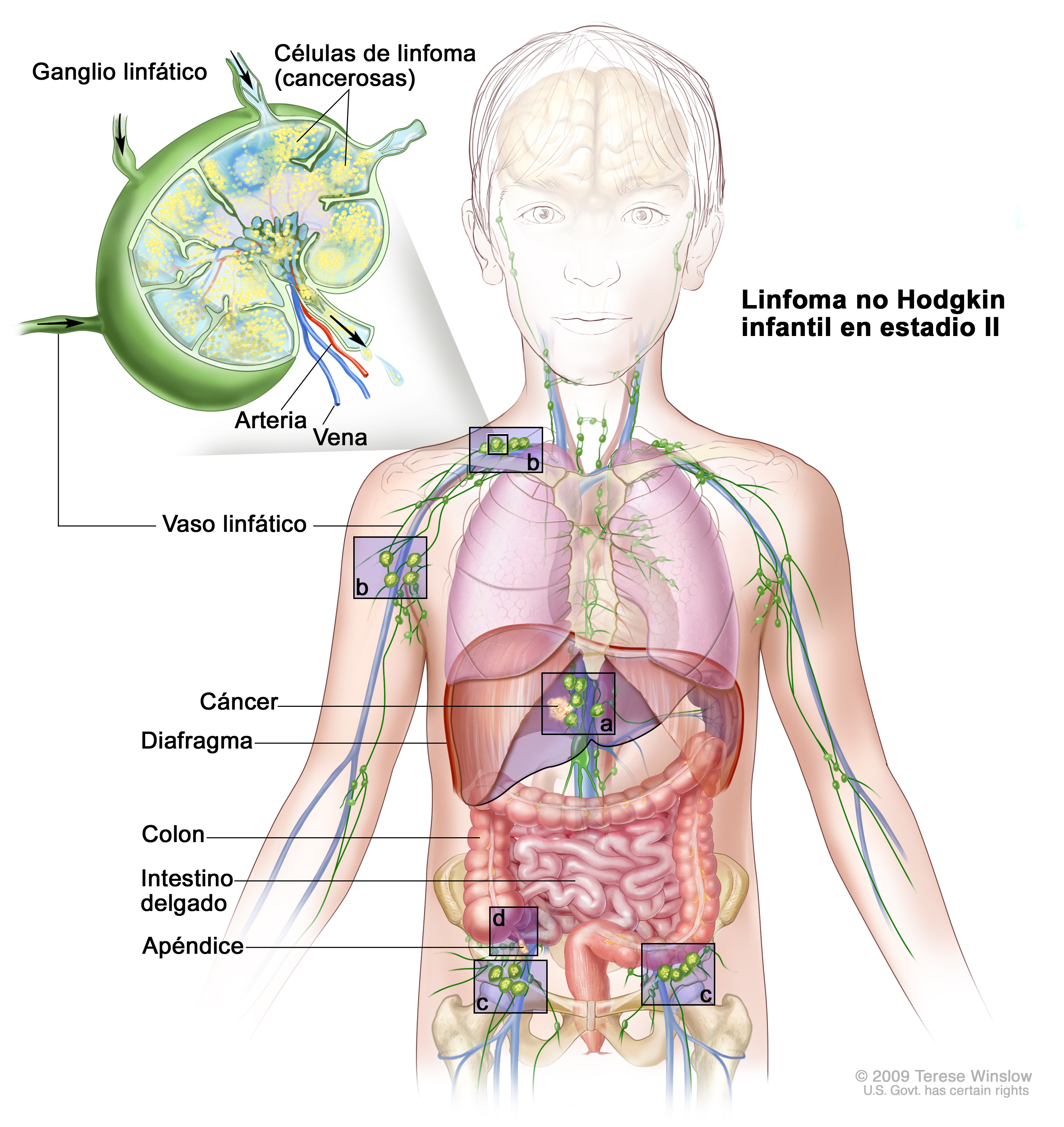
Estadio II:
Afección de 2
o más regiones ganglionares en el mismo lado del diafragma (II), o afección localizada
de un solo órgano (localización extralinfática) y su ganglio o ganglios
regionales con o sin de otras regiones ganglionares en el mismo lado del
diafragma (IIE).
Estadio III:
Afección
ganglionar en ambos lados del diafragma (III), que puede acompañarse de:
- Afección
localizada de un órgano (IIIE), o
- Localización
extralinfática asociada (IIIE), con afección del bazo (IIIs), o ambas (IIIE+s).

Estadio IV:
Afección
diseminada (multifocal) de uno o más órganos extralinfáticos, con o sin
afección ganglionar asociada, o afección extralinfática aislada con afección
ganglionar a distancia (no regional). Incluye compromiso de hígado, médula,
pulmones o líquido cerebroespinal.

III. SÍNTOMAS Y SIGNOS
Los síntomas y signos se
relacionan principalmente con la localización, el número y la extensión de las
masas ganglionares implicadas. La mayoría de los pacientes se presentan con
adenopatías cervicales y mediastínicas, pero sin síntomas sistémicos. A medida
que la enfermedad se disemina por el sistema mononuclear fagocítico,
generalmente a localizaciones contiguas, se desarrollan otras manifestaciones.
La velocidad de progresión varía
según el subtipo histopatológico. Puede aparecer precozmente un prurito
intenso; a menudo hay fiebre, sudación nocturna y pérdida de peso cuando están
afectados ganglios internos (retroperitoneal o mediastínicos voluminosos),
vísceras (hígado) o médula ósea. En ocasiones se observa fiebre de Pel-Ebstein
(algunos días de fiebre elevada que alternan regularmente con días o semanas de
temperatura normal o inferior a la normal).
Un síntoma de mecanismo poco
claro que puede aportar una clase diagnóstica precoz es el dolor inmediato en
las regiones afectadas tras ingerir bebidas alcohólicas. La afección ósea suele
ser asintomática, pero puede producir dolor con lesiones osteblásticas
vertebrales (vértebras de «marfil») y, raras veces, lesiones osteolíticas con
fracturas por compresión. La pancitopenia se debe en ocasiones a la invasión de
la médula ósea, en general en la variedad de depleción linfocítica. La invasión
epidural que comprime la médula espinal puede ocasionar paraplejía. El síndrome
de Horner y la parálisis laríngea pueden ser el resultado de la presión
ejercida por los ganglios linfáticos aumentados de tamaño sobre los nervios
simpáticos cervicales y recurrentes laríngeo, respectivamente.
Los dolores neurálgicos son
consecuencia de la compresión de las raíces nerviosas. Raras veces aparecen
lesiones intracraneales, gástricas y cutáneas y, en caso de estar presentes,
sugieren enfermedad de Hodgkin asociada al VIH.
La obstrucción de los conductos
biliares intrahepáticos o extrahepáticos por masas tumorales produce ictericia.
El edema en las piernas puede ser consecuencia de la obstrucción linfática en
la pelvis o la ingle. La compresión traqueobronquial puede causar disnea
intensa y sibilancias. La infiltración del parénquima pulmonar puede simular
una consolidación lobular o una bronconeumonía y originar cavitación o abscesos
pulmonares.
La mayoría de los pacientes
presenta un trastorno lentamente progresivo de la inmunidad retardada o
celular, que contribuye en la enfermedad avanzada a la aparición de infecciones
bacterianas frecuentes y, más raramente, de infecciones por hongos, virus y
protozoos.
La inmunidad humoral o función de
las células B también está deprimida en la enfermedad avanzada. La caquexia es
habitual y los pacientes fallecen frecuentemente por sepsis.
IV. DIAGNÓSTICO
El diagnóstico de la enfermedad
de Hodgkin es anatomopatológico y debe realizarse con las técnicas habituales y
con el inmunofenotipo para clasificar las distintas formas histopatológicas. El
diagnóstico diferencial se establece prioritariamente con los LNH. El linfoma B
mediastínico tiene células que se asemejan a las de Reed-Sternberg, los
linfomas anaplásicos pueden confundirse con la enfermedad de Hodgkin tipo
depleción linfocitaria, y la enfermedad de Hodgkin tipo predominio linfocitario
se considera en la actualidad como un linfoma de fenotipo B. Otras enfermedades
que pueden plantear problemas de diagnóstico diferencial con la enfermedad de
Hodgkin son las adenopatías de tumor primario desconocido, la
mononucleosis, sarcoidosis, la toxoplasmosis
y el síndrome de Sjögren. En la tinción de May-Grünwald-Giemsa los nucléolos
aparecen azulados, confiriendo a la célula un aspecto muy característico. En la
práctica sólo se halla como elemento gigante predominante en 1 de cada 5
pacientes, por lo que es mucho más frecuente que dominen las otras variantes de
célula de Reed-Sternberg.

V. TRATAMIENTO
Los avances terapéuticos en la
enfermedad de Hodgkin permiten en el momento actual una esperanza de curación a
20 años del 75% de los pacientes, con una toxicidad cada vez menor en cuanto a
la quimioterapia y a la radioterapia y una reducida morbilidad al ser muy
limitadas las indicaciones de la laparotomía.
Radioterapia
Representó la
primera posibilidad de curación de la enfermedad de Hodgkin en sus formas localizadas.
La aplicación de la radioterapia se realiza en zonas afectadas y adyacentes. La
erradicación del tumor depende de la dosis y se necesitan 35-40 Gy en los
lugares invadidos y 30-35 Gy en las zonas clínicamente no afectadas,
administrados a una dosis de 1,5-2 Gy/día durante 5 días a la semana.
Quimioterapia
La combinación de agentes alquilantes y de
alcaloides de la Vinca proporcionan un número importante de respuestas
completas (65%).
El MOPP (mecloretamina, vincristina, procarbazina,
prednisona), introducido en 1967, permitió un 80% de respuestas completas con
la mitad de curaciones. Sobre el protocolo MOPP se han introducido diversas
variables con la finalidad de intentar mejorar su eficacia o reducir su
toxicidad. En la actualidad no se utiliza al haberse suprimido la
mecloretamina.
El protocolo ABVD (adriamicina, bleomicina, vinblastina,
dacarbazina) tiene como base el sinergismo de sus fármacos, la eficacia de las
antraciclinas en linfomas y la utilización de fármacos no contenidos en el
programa MOPP. Sobre esta base se combinaron ambos tratamientos de forma
alternativa.